BioCantor Annotation Data Structures
The above object paradigm is used to construct a hierarchical set of objects that encapsulate common genomic concepts, and associated interval arithmetic operations.
The core data structure is biocantor.gene.feature.AbstractFeatureInterval, which has two child classes:
biocantor.gene.transcript.TranscriptInterval are used to model transcribed features. They can be coding or non-coding.
In contrast, biocantor.gene.feature.FeatureInterval are intended to be generic intervals, which may be transcribed or non-transcribed.
Both of these intervals implement a variety of methods that allow for operations such as:
Coordinate translations between genomic, CDS and transcript coordinate systems. These operations can be performed as either points or as intervals.
Sequence extraction in all possible coordinate systems.
Export to GFF3, BED and JSON.
Additionally, accessory modules exist to export groups of intervals to GenBank and NCBI TBL format, as well as helper functions to easily produce GFF3 with embedded FASTA.
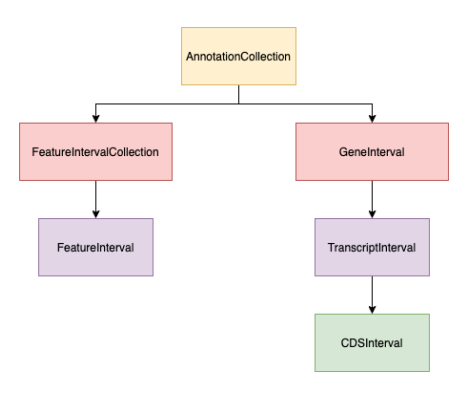
Both transcripts and features have container classes, called biocantor.gene.collections.GeneInterval
and biocantor.gene.collections.FeatureIntervalCollection respectively. These classes contain one or more
of their constituent types, and provide methods for iterating over the members.
The final core data model is biocantor.gene.collections.AnnotationCollection. This object contains one or more
GeneInterval and FeatureIntervalCollection.
This is intended to model an arbitrary genomic interval on a single sequence/chromosome. This object contains
methods that allow a user to subquery the interval by position or by identifiers of constituent objects. This object
is produced when parsing annotation file formats.
Interval Hierarchies
All interval types in BioCantor can be arbitrarily joined (“spliced”), including overlapping intervals and 0-bp gaps,
except for biocantor.gene.collections.AnnotationCollection, which are always unjoined intervals representing
the bounds of their constituents, the bounds of the sequence they derived from, or the bounds of their subquery,
depending on the source of the object.
Genes
BioCantor models the concept of genes as a three layer hierarchy:
GeneInterval -> TranscriptInterval -> CDSInterval (optional)
Under this model, a gene can be considered to be any sort of transcribed interval. This is different from the Sequence Ontology model, where transcribed intervals as well as non-transcribed intervals are direct children of the biological_region term. This model is a simplification that can have issues – for example, pseudogenes that are not transcribed.
All biocantor.gene.transcript.TranscriptInterval and biocantor.gene.collections.GeneInterval
objects have an optional biocantor.gene.biotype.Biotype associated that assigns a INSDC-style Biotype
to the interval. TranscriptInterval objects do not have to have the same biotype
as their parent gene. This follows the GENCODE/Ensembl model for biotype assignment, rather than the NCBI model.
Non-transcribed intervals (Features)
BioCantor models the concept of generic intervals as a two layer hierarchy:
FeatureIntervalCollection -> FeatureInterval
This is intended to allow for grouping of non-transcribed features. An example of this could be a promoter region,
with multple known transcription factor binding sites. In this example, the promoter could be built as a
FeatureIntervalCollection, with a child
FeatureInterval for each TFBS.
While both of these objects do allow for joined/compound intervals, doing so is of course optional and not as likely to make sense for many use cases.
Unlike genes, FeatureInterval do not have a restricted ontology of types. Additionally,
they can have multiple types, stored as the value feature_types. When a
FeatureIntervalCollection is constructed, it adopts the union of all types
of its children FeatureInterval. This allows for set operations to occur - as in
the above example, there could be three intervals with types ["tfA", "tfB", "tfC"] respectively, and thus the
collection containing them will have all three of those types associated with it.
FeatureIntervalCollection also can have one type of its own, which in the above
example could be promoter.